 华商检测认证机构中心
华商检测认证机构中心【今日观点】 哪些食品申请FDA认证属于酸性罐头类?
美国食品和药物管理局要求生产某些酸化和低酸罐装食品(LACF)包装在密封容器,以获得食品罐头公司(FCE)注册。此外,食品罐头公司必须向食品和药物管理局提交文件,说明在生产符合这些要求的食品时所使用的





美国食品和药物管理局要求生产某些酸化和低酸罐装食品(LACF)包装在密封容器,以获得食品罐头公司(FCE)注册。此外,食品罐头公司必须向食品和药物管理局提交文件,说明在生产符合这些要求的食品时所使用的
美国食品药品监督管理局(US FDA)负责确保在美国销售的食品安全,有益健康并贴有适当标签。这适用于国内生产的食品,以及国外的食品。联邦食品,药品和化妆品法(FD和C法)以及《公平包装和标签法》是管辖
美国食品药品监督管理局(FDA)化妆品注册系统于2023年12月16日正式开放!自2022年12月29日,美国颁发《2022年化妆品法规现代化法案》(MoCRA)以来,FDA一直在不断完善注册要求及新
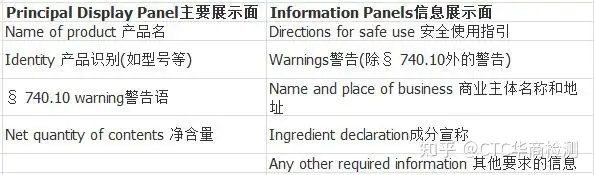
FDA《商品包装和标签法》的颁布其实就是对化妆品标签的严格而明确的规定。在市场上销售的化妆品都必须要符合这个规定的,因为这是评价化妆品合格与否的主要依据,也是管理化妆品的主要内容,标签不符合要求的化妆
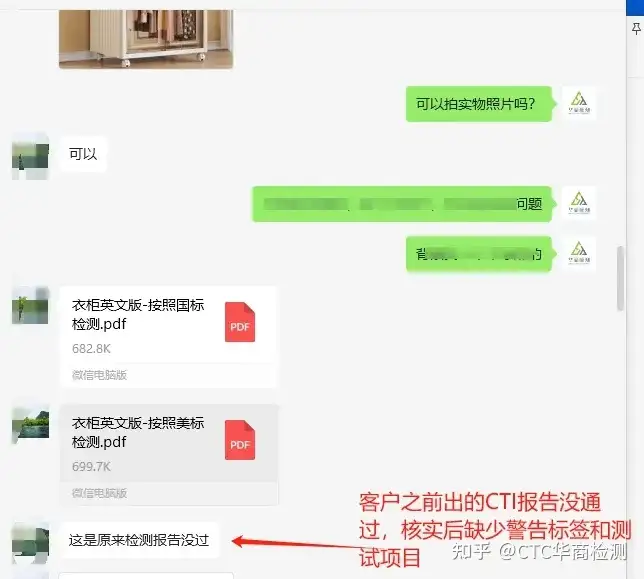
2023 年 4 月 19 日,美国消费品安全委员会投票通过 ASTM F2057-23 作为强制性安全标准,以防止梳妆台和其他衣物存储单元等家具翻倒而造成幼儿受伤害和死亡。2022年12月STURD
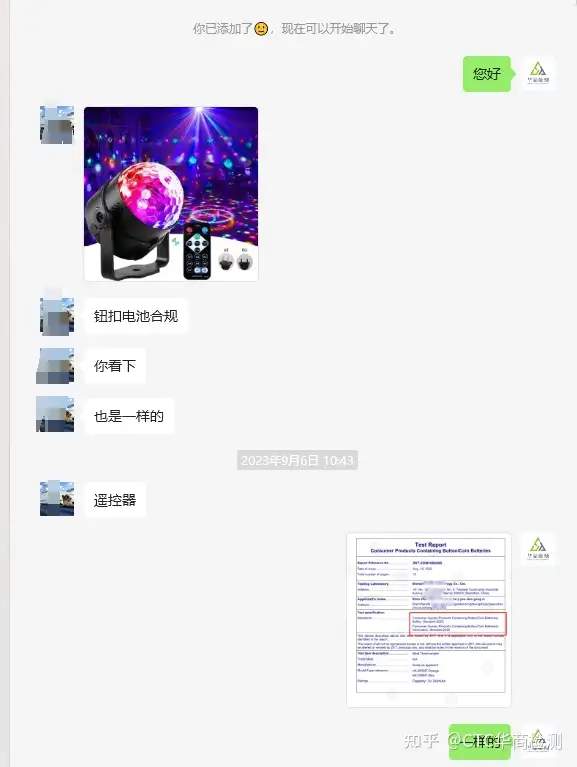
近日,亚马逊美国站公布要求卖家需遵守扭电池和硬币电池的新包装和警示标签规定公告。在亚马逊销售单独的纽扣电池和硬币电池,则从2023年3月2日开始,您需要证明您的符合儿童安全包装和警告标签要求。适用产品

婴儿学步车是一种移动装置,当坐在或站立在学步车内的儿童推动时,该装置可使儿童在水平表面上移动。产品示例婴儿学步车标准的目的是什么?该标准旨在最大限度减少与使用婴儿学步车有关的死伤风险, 包括与摔下楼梯

美国联邦法律规定,地毯和垫子要符合易燃性标准和其它要求, 包括2008年《美国消费品安全改进法》的要求。在地毯和垫子经过检测或合理检测项目后,作为一般用途的 地毯和垫子的生产商和进口商必须在一般合规证

美国消费品安全委员会CPSC于2023年9月26日在联邦公报上发布了哺乳枕新法规16 CFR 1242的草案,旨在降低哺乳枕使用带来的伤害和死亡风险。该法规草案提到了在使用哺乳枕时由于婴儿睡着或无人看
乌克兰新的认证制度-乌克兰TR认证(也叫乌克兰技术法规声明)英文简称:Technical Regulations (Declaration),俄文:техническим регламентам (д
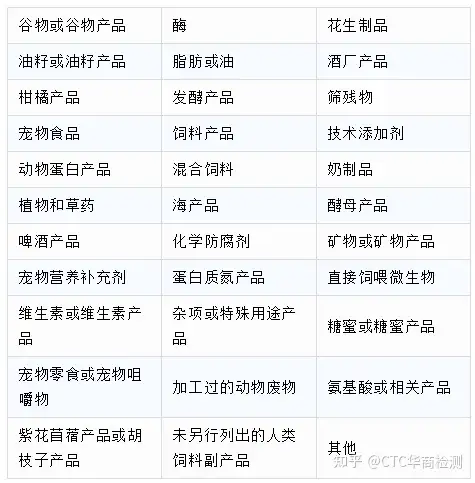
根据美国FDA规定,对于进入美国市场的饲料类产品必须要对其生产企业或公司信息在美国FDA官网完成FDA注册登记,获得相关FDA注册号方可通关。美国食品和药物管理局 (FDA) 负责监管猫粮、狗粮以及零
FDA(美国食品和药物管理局Food and Drug Administration)对医疗器械的管理通过器械与放射健康中心(CDRH)进行的,中心监督医疗器械的生产、包装、经销商遵守法律下进行经营活

随着国内企业日益壮大,所以现在国内企业都将目光放到了海外市场,尤其是欧美市场。但想要将自己产品出口到海外市场有一个很大的问题,那么医疗器械进入美国市场需要做FDA,那FDA认证如何查询分类呢?我们一起
上篇文章我们讲解了激光产品美国FDA认证注册要求及办理流程(点击查看)今天我们主要讲解一下激光产品FDA/CDRH登录认证注册号是什么?为什么激光产品需要注册FDA认证?首先激光类产品为什么要做FDA
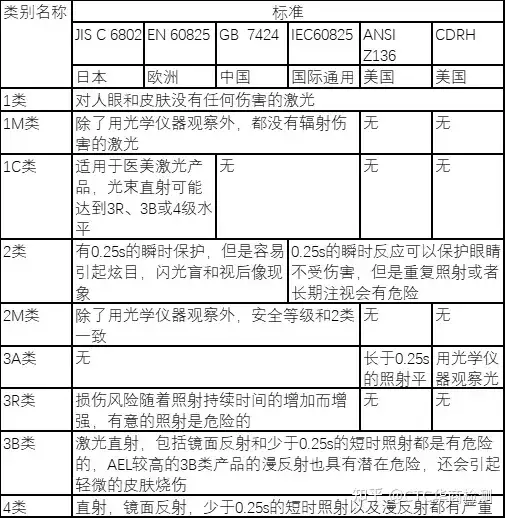
激光主要危害视网膜灼伤、眼部病变、皮肤烧伤、火灾、光化学反映危害、有毒粉尘危害、电灯。为了确保激光设备的安全性能,国际电工委员会(InternationalElectrotechnicalCommis

近期收到很多做美国亚马逊的卖家咨询,大多数都是店铺里正在销售的产品突然被下架, 要求提供21 CFR1040.10认证或者IEC60825认证,上篇文章我们讲解了激光产品安全标准IEC60825-1如

DMF(Drug Master File,药品文献档案)是美国FDA要求的一种非公开文件,用于提交药品、原料药、包装材料、助剂、配方和工艺等有关数据的注册文件。DMF通常由药品原料、药品包装和助剂的制

俄罗斯的医疗器械产品注册由卫生部(Roszdravnadzor,或 RZN)控制。按照俄罗斯联邦政府于 2012 年 12 月 27 日颁布的第 1416 号法令要求的方式注册的医疗用途产品才允许在俄
510K指的是FDA法规里面的一个章节,讲的是PMN,也就是Pre Market Notification 上市前通告。510(k)文件是向FDA递交的上市前申请文件,目的是证明申请上市的器械与不受上
医疗器械510k和GMP豁免范围介绍:合规文件外,只有带有(注释的设备也免于 GMP以下是按设备类别列出的 510k豁免和良好生产规范(GMP)/质量体系豁免的细分,除非通过脚注进一步限定,否则此列表
2017年4月,欧盟医疗器械法规(MDR)和体外诊断医疗器械法规(IVDR)发布,法规中定义了欧盟UDI系统的相关要求此外,可通过该标识对欧盟医疗器械数据库(EUDAMED)中的医疗器械进行分组,为了
医疗器械法规 – MDR (EU 2017/745) 和体外诊断法规 – IVDR (EU 2017/746) 要求公司配备一名监管合规负责人 (PRRC)。 PRRC 是一位监管专家,其职责是确保公

ANVISA,全称Agência N.acional de Vigilancia Sanitária,隶属巴西卫生部,负责所有医疗器械、体外诊断产品及其他健康相关产品(如药品、卫生用品、化妆品等)的上
 韩国KC认证证书怎么查询?2020-11-16
韩国KC认证证书怎么查询?2020-11-16
 IEC和EN标准有什么区别?2020-11-11
IEC和EN标准有什么区别?2020-11-11
 倍科(BACL)被查已8年伪造报告获取KC认证1700例,涉及381家公司包括华为,DJI, 海康等2020-11-16
倍科(BACL)被查已8年伪造报告获取KC认证1700例,涉及381家公司包括华为,DJI, 海康等2020-11-16
 CCC认证查询的几种详细方法2020-10-27
CCC认证查询的几种详细方法2020-10-27
 谷歌GMS认证CTS,GTS测试介绍2020-10-29
谷歌GMS认证CTS,GTS测试介绍2020-10-29
 市面上目前有哪些苹果数据线有过MFI认证呢?2020-11-09
市面上目前有哪些苹果数据线有过MFI认证呢?2020-11-09
 EN/IEC62311:2020更新_关于人体电磁场暴露测试要求2020-11-12
EN/IEC62311:2020更新_关于人体电磁场暴露测试要求2020-11-12
 未办理CCC认证怎么处罚?2020-10-27
未办理CCC认证怎么处罚?2020-10-27
 CTA入网电信设备进网常见问题解答!2020-10-29
CTA入网电信设备进网常见问题解答!2020-10-29
 什么是中国CCC认证?2020-10-27
什么是中国CCC认证?2020-10-27
 什么是谷歌GMS认证?2020-10-29
什么是谷歌GMS认证?2020-10-29
 谷歌GMS认证过程问题分析2020-10-29
谷歌GMS认证过程问题分析2020-10-29
